Table of contents
Gene finders list
RNAspace userguide
July 2011
Table of contents
Introduction
RNAspace is a web platform to support non-protein-coding RNA (ncRNA)
identification on genomic sequences. The platform allows running in
an integrated environment a variety of
ncRNA gene finders, to explore results with
dedicated tools for comparison, visualization and edition of putative
ncRNAs and to export them in various formats (
FASTA,
GFF,
CSV,
RNAML).
The environment is developed as a collaboration between several French bioinformatics platforms and laboratories.
The environment is developed as a collaboration between several French bioinformatics platforms and laboratories.
It is an Open Source software available through:
- a website http://rnaspace.org with a selection of annotation tools and with limitations on the genomic sequence size to be analyzed and on execution time,
- local installation on computer platforms with user authentication.
Requirements.The RNAspace website requires JavaScript and CSS
(Cascading Style Sheets). The following browsers are supported:
Mozilla Firefox, Google Chrome, Microsoft Internet Explorer 7 and
8. For the latter, just switch Compatibility View on
using the toolbar button or menu option to have a correct
visualization of chosen organisms in the Predict page.
Navigation on rnaspace.org
All pages have the same header, which features four tabs:

The Home tab leads to RNAspace
home page. The three other tabs are used to perform analyses of genomic
sequences.
- Home
- Load data
- Predict
- Explore
The first step is to access the Load page, using the Load
data tab, and to enter the query sequences. This starts a new
project.
When this is done, the Predict page is displayed. There, in the second step,
you have to choose a set of annotation tools,
and start the computation. This starts the first run of the current
project: A run is the execution of a selection of gene finders on your data.
A 6-letter identifier (ID) is associated to the project, and the run is
numbered r01.
Once results are obtained, the Export page is automatically displayed,
allowing you to examine predicted putative RNAs, modify them,
export them. At this third step, you can also return to the
Predict page using the Predict tab to select additional program
tools on the same data, or same programs with modified parameter values.
This starts a new run in the current project.
If you want to analyze new data, you have to proceed to the Load
page using the Load data tab. By doing this, you start a new project,
and the previously uploaded data and associated results are lost.
All pages feature a help button in the top right of the
page. It links to this context-sensitive Help page, which is opened in a
new tab (or a new window, depending on your browser setting).
This tab is opened just once during the session.
Load data page: load genomic sequences
In this page, you have to load the genomic sequences you want to
analyze and give some additional information on each sequence, such as the domain, the species,...
Enter a query sequence.
You can either paste the sequence in the text area or upload it
from a local file. In both cases, the FASTA format is required.
FASTA format consists of a comment line (a line beginning with '>')
followed by the genomic sequence. The sequence should contain only A, U,
T, G, C, N letters in either upper or lower case.
There is a global size limitation on loaded sequences: 5.0 Mb nucleotides.
> sequence 1
ACGUGCUCGUACGUGCACGUUGCUCCG
CGUUUGUAGCUGUUGAGUGAGCCGAUG
Note that a multiFASTA file, that is a concatenation of sequences
in FASTA format, is accepted when sequences share the same values
for required information (see below). The number of sequences per
multiFASTA is limited to 20 sequences.There is a global size limitation on loaded sequences: 5.0 Mb nucleotides.
Enter the sequence name. By default, a name of the
form seq_000001 is given. The name must be made up of upper
and lower case letters, figures and '_', '.', '-' characters.
The length of the name must be between 1 and 40 characters.
Specify the domain of the sequence.
There are three possible domains: Bacteria, Archaea and Eukaryote.
This information will be useful to provide suitable prediction tools
in the Predict step.
Optionally, you can also specify:
- the species, such as Escherichia_coli,
- the strain, such as K12,
- the replicon, such as chromosome or plasmid1.
Click on the upload button to upload the sequence. The
format of the sequence and all given values are then checked.
If the control is successful, the sequence is added to your data and
displayed in a summary table.
You can enter as many sequences as you want by repeating this process,
provided that you do not exceed the total limitation
of 5.0 Mb nucleotides.
Once all sequences are loaded, you should enter your Email
address (multiple addresses splited by a coma), in order to be
alerted when computation will be finished. You can then access the Predict
page by clicking on the submit button, at the bottom of the page.
Predict page: choose gene finders
RNAspace offers you a collection of complementary gene finders to
help identify ncRNAs.
Select one or several tools by ticking the corresponding check box.
For each tool, a short description can be accessed by clicking on the
associated 'more' hyperlink. The '?' hyperlink, at the end of the
description, refers to this general Help page and displays the related
section.
Optionally modify parameters value.
Each gene finder is proposed with default parameter values.
There are two kinds of parameters: visible ones, displayed on the
Predict page (in drop-down lists), and advanced ones accessible by
clicking on the parameters button to the right on the gene finder
name. All gene finders do not have visible and/or advanced parameters.
Be careful: parameters are modified for all active Predict pages
of the current session.
For the sake of clarity, available annotation tools are organized in
three sections.
- Homology search based gene finders
- Comparative analyzis based gene finders
- ab initio gene finders
Time limitation on rnaspace.org. Maximum allowed
running time for a gene finder is 8 hours. If the computation
exceeds this duration, then the process is stopped.
Homology search based gene finders
Sequence homology search tools
RNAspace first proposes sequence search against ncRNA
databases. The following databases are available.
- Rfam is a collection of multiple sequence alignments and covariance models covering many ncRNA families. RNAspace currently proposes Rfam 9.1 (Dec. 2008, 1371 families) [more information on RFAM 9.1] and Rfam 10.0 (Jan. 2010, 1446 families) [more information on RFAM 10.0].
- fRNAdb is a database of comprehensive ncRNA sequences including known (or previously reported) sequences, which are acquired from other databases (FANTOM3,H-invDB 5.0, miRBase 10.0, NONCODE 1.0, Rfam 8.1, RNAdb 2.0, snoRNA-LBME-db 3, GEO), and sequences reported by the joint research groups of the Functional RNA Project [more information on fRNAdb].
- miRBase 13.0 is database of published miRNA sequences and annotation. RNAspace currently uses miRBase 13.0 (March 2009), which contains 9539 entries [more information on miRBase].
When comparing the query sequence against a ncRNA database, all
alignments referring to the same regions for a same family are
merged in a single prediction. Only best alignments are stored
(the number of stored alignments can be modified in advanced parameter
section).
There are several choices of alignment tools for database search.
-
BLAST finds regions of local similarity between sequences using k-mers.ApproachBLAST searches for high scoring sequence alignments between the query sequence and sequences in the database using a heuristic approach that approximates the Smith-Waterman algorithm for local alignment. The main idea of BLAST is that there are often high-scoring segment pairs (HSP) contained in a statistically significant alignment. BLAST first search for these HSPs, then extend them to construct valid alignments.
RNAspace currently uses BlastN 2.2.25.
ParametersDatabase: The ncRNA database to which the query sequence will be compared.
Number of alignments: When comparing the request sequence to the reference database, any putative RNA can match many homologous ncRNAs occurring in the same family in the database. Such overlapping alignments are merged into a single prediction. This parameter specifies the maximal number of pairwise alignments that will be saved for each putative ncRNA. Selected alignments are sorted according to their E-value. Default value is 5.
Filter low complexity regions in the query sequence: This function masks off segments of the query sequence that have low compositional complexity, as determined by the DUST program of Tatusov and Lipman. Filtering can eliminate statistically significant but biologically uninteresting reports from the BLAST output. Filtering is only applied to the query sequence, not to database sequences. Default value is yes.
When this option is selected, the complementary option "Mask for lookup table" is automatically set to true. It means that no HSPs are found based upon low-complexity sequence, but the BLAST extensions are performed without masking and so they can be extended through low-complexity sequence.
Query strand : The comparison of the query sequence against the database can be performed on the forward strand only, the reverse strand only, or on both strands. Default value is both.
Word size (between 7 and 30): This is the size of the HSPs (or k-mers) used in BLAST heuristics to identify potential high scoring alignments. One normally regulates the sensitivity and speed of the search by increasing or decreasing the word size. The lower is this value, the more sensitive is the algorithm. The higher is this value, the faster is the algorithm.
Match/mismatch/gap opening/gap extension scores: This is simply the scoring system used to assess the quality of a pairwise alignment. In BLAST, there are internal dependencies between these score parameters. So they are available in a pull down menu that shows the allowed reward/penalty pairs and their associated gap existence and gap extension penalties. Default values are 2 for a match, -3 for a mismatch, 5 for a gap opening, and 2 for a gap extension.
E-value (expectation value) threshold: This setting specifies the statistical significance threshold for reporting alignments against database sequences. For example, when this parameter is set to 5, it means that 5 such matches are expected to be found merely by chance, according to the stochastic model of Karlin and Altschul (1990). Lower E-value thresholds are more stringent, leading to fewer chance matches being reported.
Score of RNAspace predictionsWhen several alignments are involved in a prediction, RNAspace reports the best E-value (the lowest E-value) as the score of the prediction.
ReferencesAltschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. (1990) "Basic local alignment search tool." J. Mol. Biol. 215:403-410.
NCBI BLAST: a better web interface Mark Johnson, Irena Zaretskaya, Yan Raytselis, Yuri Merezhuk, Scott McGinnis, and Thomas L. Madden Nucleic Acids Res. 2008 July 1; 36(Web Server issue): W5-W9.
BLAST NCBI web site -
YASS finds regions of local similarity between sequences using spaced-seeds.ApproachLike most of the heuristic DNA local alignment software, YASS uses seeds to detect potential similarity regions, and then tries to extend them to actual alignments. It has the distinctive feature of using multiple transition-constrained spaced seeds to ensure a good sensitivity/selectivity trade-off. That enables to search more fuzzy conserved sequences, as non-coding RNAs.
Spaced seed is a non-contiguous pattern, that allows for some errors. Transition mutations are purine to purine [A<->G] or pyrimidine to pyrimidine [C<->T].
RNAspace currently uses YASS 1.14.
ParametersDatabase: The ncRNA database to which the query sequence will be compared.
Number of alignments: When comparing the request sequence to the reference database, any putative RNA can match many homologous ncRNAs occurring in the same family in the database. Such overlapping alignments are merged into a single prediction. This parameter specifies the maximal number of pairwise alignments that will be saved for each putative ncRNA. Selected alignments are sorted according to their E-value. Default value is 5.
Filter low complexity regions in the query sequence : This function masks off segments of the query sequence that have low compositional complexity. Filtering can eliminate statistically significant but biologically uninteresting reports from the YASS output. Filtering is only applied to the query sequence, not to database sequences. Default value is yes.
Query strand : The comparison of the query sequence against the database can be performed on the forward strand only, the reverse strand only, or on both strands. Default value is both.
Seed Pattern(s): You can specify the seed pattern used in the search step. The "\#" symbol is for matches, that is positions without error. The "\@" symbol is for matches or transitions, and the "_" is for any possibility; match, transition or transversion. The comma "," can be used as a seed separator. The program speed depends on the weight of each pattern (number of # + 1/2 number of @): decreasing the weight increases sensitivity, but slows down the search.
Seed hit criterion: you can specify the number of seeds that have to match the query sequence. Default value is double.
Match/transversion/transition/undetermined symbol scores: This is simply the scoring system used to assess the quality of a pairwise alignment. Default values are 5 for a match,-4 for a transversion, -3 for a transition and -4 for undetermined symbols.
Gap costs: This is the affine penalty scores associated to gaps in the score of a pairwise alignment. Default values are -16 for a gap opening and -4 for a gap extension.
E-value (expectation value) threshold: This setting specifies the statistical significance threshold for reporting alignments against database sequences. For example, when this parameter is set to 5, it means that 5 such matches are expected to be found merely by chance, according to the stochastic model of Karlin and Altschul (1990). Lower E-value thresholds are more stringent, leading to fewer chance matches being reported.
Score of RNAspace predictionsWhen several alignments are involved in a prediction, RNAspace reports the best E-value (the lowest E-value) as the score of the prediction.
ReferenceNoe L., Kucherov G. YASS: enhancing the sensitivity of DNA similarity search Nucleic Acids Research, 33(2):W540-W543, 2005.
YASS web site
General purpose RNA motif
search tools
These programs use homology both at the sequence level and at the
structure level to identify potential occurrences of ncRNAs.
Each program comes with its own compilation of RNA profiles.
-
Darn provides a wide-range of RNA profiles. The search algorithm is based on constraint networks.ApproachThe problem of finding new occurrences of characterized ncRNAs can be modeled as the problem of finding all locally-optimal solutions of a weighted constraint network using dedicated weighted global constraints, encapsulating pattern-matching algorithms and data structures. This is embodied in Darn, a software tool for ncRNA localization, which, compared to existing pattern-matching based tools, offers additional expressivity (such as enabling RNA---RNA interactions to be described) and improved specificity (through the exploitation of scores and local optimality) without compromises in CPU efficiency.
RNAspace currently uses version 1.0.
ParametersDescriptor: Descriptor of searched RNA family. Available descriptors are provided in a list.
The first element of the list is 'All [domain]'. This allows to select all descriptors suitable to the request sequence domain.
The others lines describes a RNA family name followed by the suitable domain(s) for the family. The notation is 'B' for Bacteria, 'A' for Archaea, 'E' for Eukaryote, separated by '/' and surrounded by squared bracket. For example 'RNaseP [B/A/E]' selects a descriptor of RNaseP suitable for all domains.
Note that it is not forbidden to try a descriptor not suitable for the request sequence domain but user will have to keep it in mind when analysing results of prediction.
Score of RNAspace predictionsNo score for predictions (set at zero).
ReferenceDARN! A Weighted Constraint Solver for RNA Motif Localization.
Matthias Zytnicki, Christine Gaspin, Thomas Schiex. Constraints, Volume 13 (2008). -
ERPIN provides a wide-range of RNA profiles. The search algorithm is based on dynamic programming.ApproachERPIN (Easy RNA Profile IdentificatioN) is an RNA motif search program. Unlike most RNA pattern matching programs, ERPIN does not require users to write complex descriptors before starting a search. Instead ERPIN reads a sequence alignment and secondary structure, and automatically infers a statistical "secondary structure profile" (SSP). An original Dynamic Programming algorithm then matches this SSP onto any target database, finding solutions and their associated scores.
RNAspace currently uses version 9.1.
ParametersTraining set: Descriptor of a RNA family, The first element of the list is 'All [domain]'. This allows to select all descriptors suitable to the request sequence domain.
The others lines describes a RNA family name followed by the suitable domain(s) for the family. The notation is 'B' for Bacteria, 'A' for Archaea, 'E' for Eukaryote, 'V' for Virus, separated by '/' and surrounded by squared bracket. For example 'Bacterial 5S rRNA [B]' selects a descriptor of Bacterial 5S rRNA suitable for Bacteria domains.
Note that it is not forbidden to try a descriptor not suitable for the request sequence domain but user will have to keep it in mind when analysing results of prediction.
Score of RNAspace predictionsNo score for predictions (set at zero).
ReferenceGautheret D, Lambert A. Direct RNA Motif Definition and Identification from Multiple Sequence Alignments using Secondary Structure Profiles. J Mol Biol. 313:1003-11 (2001).
ERPIN web site -
INFERNAL is for searching DNA sequence for RNA structure and sequence similarities. Here it uses family descriptors (covariance models) provided by Rfam 10.0 database.ApproachINFERNAL ("INFERence of RNA ALignment") is for searching DNA sequence databases for RNA structure and sequence similarities. It is an implementation of a special case of profile stochastic context-free grammars called covariance models (CMs). A CM is like a sequence profile, but it scores a combination of sequence consensus and RNA secondary structure consensus, so in many cases, it is more capable of identifying RNA homologs that conserve their secondary structure more than their primary sequence.
RNAspace uses Infernal 1.0.2 and a subset of Rfam 10.0 models.
The models related to the following falilies were ignored: miRNAs, rRNA (except 5.8S), tRNA, IRES, CRISPR, Intron, RF.
ParametersDescriptor: Descriptor of searched RNA family. Available descriptors are provided in a list. The 'All' descriptor corresponds to all defined descriptors (1446 families).
Note that it is not forbidden to try a descriptor not suitable for the request sequence domain but user will have to keep it in mind when analysing results of prediction.
Score of RNAspace predictionsThe score of a prediction is the Infernal E-value (see the user guide for more information).
ReferenceE. P. Nawrocki, D. L. Kolbe, and S. R. Eddy, Infernal 1.0: Inference of RNA alignments , Bioinformatics 25:1335-1337 (2009).
INFERNAL web site
Specialized search tools
Each program is dedicated to a family of ncRNAs.
-
RNAmmer predicts ribosomal RNA genes (5s/8s, 16s/18s, and 23s/28s).ApproachRNAmmer predicts ribosomal RNA genes in full genome sequences by utilising two levels of Hidden Markov Models: An initial spotter model searches both strands. The spotter model is constructed from highly conserved loci within a structural alignment of known rRNA sequences. Once the spotter model detects an approximate position of a gene, flanking regions are extracted and parsed to the full model which matches the entire gene. By enabling a two-level approach it is avoided to run a full model through an entire genome sequence allowing faster predictions.
RNAspace currently uses version 1.2.
ParametersSearch for 5/8S rRNA: Set RNAmmer to search for 5/8S rRNA.
Search for 16/18S rRNA: Set RNAmmer to search for 16/18S rRNA.
Search for 23/28S rRNA: Set RNAmmer to search for 23/28S rRNA.
If there is no options checked, RNAmmer is settled to search for all of them.
Score of RNAspace predictionsThe score of a prediction is the RNAmmer score (see RNAmmer description for more information).
ReferenceRNammer: consistent annotation of rRNA genes in genomic sequences.
Lagesen K, Hallin PF, Rødland E, Stærfeldt HH, Rognes T Ussery DW. Nucleic Acids Res. 2007.
RNAmmer web site -
tRNAscan-SE predicts transfer RNAs.ApproachtRNAscan-SE combines the strengths of three independent tRNA prediction programs by negotiating the flow of information between them, performing a limited amount of post-processing, and outputting the results in one of several formats.
RNAspace currently uses version 1.23.
ParametersSearch using Cove analysis only (max sensitivity, very slow): Set tRNAscan-SE to detect tRNA using only the Cove analysis (neither tRNAscan nor EufindtRNA will be used). The covariance model used will depend on the sequence domain.
Check for pseudogenes: Set tRNAscan-SE to check if predicted RNAs are a pseudogene or not.
Cove cutoff score for reporting tRNAs: Set cutoff score (in bits) for reporting tRNAs (default=20)
Integer value between [10 ; 30]
Max length of (intron + variable region): Set max length of tRNA intron + variable region (default=116bp)
Integer value between [50 ; 200]
Score of RNAspace predictionsThe score of a prediction is the tRNAscan-SE score (see tRNAscan-SE description for more information).
ReferenceEddy, S.R. and Durbin, R. (1994) "RNA sequence analysis using covariance models", Nucl. Acids Res., 22, 2079-2088.
Fichant, G.A. and Burks, C. (1991) "Identifying potential tRNA genes in genomic DNA sequences", J. Mol. Biol., 220, 659-671.
Pavesi, A., Conterio, F., Bolchi, A., Dieci, G., Ottonello, S. (1994)" Identification of new eukaryotic tRNA genes in genomic DNA databases by a multistep weight matrix analysis of transcriptional control regions", Nucl. Acids Res., 22, 1247-1256.
Lowe, T.M. & Eddy, S.R. (1997) "tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence", Nucl. Acids Res., 25, 955-964.
tRNAscan-SE web site
Comparative analysis based gene finders
The rationale of comparative analysis is that ncRNAs are
under a positive selection pressure and therefore should be better
conserved than other sequences (just like coding sequences). This
gives a way to detect these conserved elements by searching for
similar sequences across species.
In the first step, you have to select at least one up to
four species to be compared with. Each species is related to
one or several genomes or strains.
In the second step, a prediction method has to be
chosen. There first a similarity search is done by either
BLAST or YASS (see description above) to collect
all conserved sequences with the query sequence. Only intergenic
regions are processed. Pairwise alignments are automatically
combined into clusters of conserved regions with CG-seq [more information on
CG-seq]. Then these conserved sequences are investigated by
inspection of evolutionary patterns to identify significantly
conserved secondary structures. RNAz and CaRNAc are available
for this task.
No score for predictions (set at zero).
No score for predictions (set at zero).
-
caRNAc predicts structurally conserved and thermodynamically stable RNA structures. It can successfully handle low similarity sequences.ApproachcaRNAc combines three features: energy minimization, phylogenetic comparison and sequence conservation. It relies on a Sankoff-like folding algorithm that simultaneously infers a potential consensus structure and align the sequences. As a consequence, the method does not require any prior multiple sequence alignment, and appears to be robust to noisy datasets.
RNAspace currently uses version 0.31.
ParametersTake GC percent into account: When this option is selected, caRNAc uses variable energy thresholds for helices selection according to the average GC percent of the involved sequence.
ReferencecaRNAc: folding families of non coding RNAs Touzet H. and Perriquet O. Nucleic Acids Research 142, 2004.
caRNAc web site -
RNAz predicts structurally conserved and thermodynamically stable RNA structures in multiple sequence alignments.ApproachRNAz combines comparative sequence analysis and structure prediction. It consists of two basic components: (i) a measure for RNA secondary structure conservation based on computing a consensus secondary structure, and (ii) a measure for thermodynamic stability, which, in the spirit of a Z-score, is normalized with respect to both sequence length and base composition. The two independent diagnostic features of structural ncRNAs are finally used to classify an alignment as "structural RNA" or "other". For this purpose, RNAz uses a support vector machine (SVM) learning algorithm which is trained on a large test set of well known ncRNAs.
RNAz predicts a consensus secondary structure for an alignment by using the RNAalifold approach. RNAalifold works almost exactly as single sequence folding algorithms (e.g. RNAfold), with the main difference that the energy model is augmented by covariance information. Compensatory mutations (e.g. a CG pair mutates to a UA pair) and consistent mutations (e.g. AU mutates to GU) give a "bonus" energy while inconsistent mutations (e.g. CG mutates to CA) yield a penalty.
RNAz requires that the data to analyse is given in a multiple sequence alignment. In RNAspace, the multiple alignment is build with ClustalW, using default parameter values for nucleic acids sequences.
RNAspace currently uses version 2.0.9.
ParametersProbability cut off: This is the probability of the sequence of being a ncRNA, as computed by the SVM. Results are displayed only for sequences having a classification probability higher than this value. Sequences with high probablity cut off (e.g.>0.9) show strong evidence for a structural RNA. Default value is 0.7.
Slice alignments longer than, Window size and Step size options: The RNAz algorithm works globally, i.e. the given alignment is scored as a whole. For long alignments (e.g alignment of a whole chromosome), this is neither computationally tractable nor biologically meaningful. Therefore, long alignments are scanned in overlapping windows. Alignments longer than the slice size will be analyzed in sliding overlapping windows of the given window size. The step size value specifies the number of shifted positions. In RNAspace, The default values are 300, 200 and 50 respectively.
ReferenceWashietl S., Hofacker I.L., Stadler P.F. Fast and reliable prediction of noncoding RNAs Proc. Natl. Acad. Sci. U.S.A. 102, 2454-2459, 2005.
RNAz web site
ab initio gene finders
The last kind of prediction tools uses intrinsical statistical feature
of the data, such as compositional bias.
-
atypicalGC identifies atypical G and C content regions in sequence with a GC% out of [40 ; 60].ApproachFirst, evaluate if the approach is appropriate, ie GC% mean on all the sequence is out of the interval [40 ; 60]. If so, on each position of a given sequence, compute G and C percent content on a sliding window centered on the considered position. Only positions with a value more distant than two standard deviation of the mean are considered as atypical.
Second, only regions (continuous atypical positions) longer than a minimum length and containing less than 5% of 'N' nucleotides are kept.
RNAspace currently uses atypicalGC 1.0.
ParametersForce prediction: force prediction even if the approach is not appropriate.
default value: 'False'
Allowed value in 'True','False'
Look for up/down/all atypical GC%: only keep regions with GC% up/down/all mean value (with sliding window), default value: 'all'
Allowed value in 'all', 'up', 'down'
Window size: sliding window size, default value: 101
odd integer value in [3 ; 501]
If non odd integer, the value is reduced by 1
Min length: minimum region length, default value: 50
Integer value between [1 ; 500]
Score of RNAspace predictionsNo score for predictions (set at zero).
ReferenceINRA BIA Toulouse, 2008.
Combine results
RNAspace allows you to launch a variety of genes finders on your data
in a single run. Of course, the usage of several tools may lead to
redundant overlapping predictions. The goal of the combine results
option is to clean up the list of results. When this option is checked,
strongly overlapping predictions having the same family name and
occurring on the same strand are merged into a single
prediction.
This is done in two steps.
-
first, create clusters of overlapping RNA prediction of the same family.
We consider that two overlapping putative RNAs must share at least 10 nucleotides and occur on the same strand.
For the family, we use an equivalent family name table, that
gives a generic name for a family and the equivalent name given by
all gene finders.
- then, for each cluster, create a new putative RNA, obtained by combination of existing predictions.
- for tRNA and rRNAs, we privilege the prediction given by specific gene finders, because these gene finders appear to give better results.
- for unknown families, a special treatment is defined because corresponding gene finders rely on so different approaches (based on sequence comparison or sequence characteristics)
- for other family names, a general default rule is defined
Here are the rules used to combine RNAs of a cluster
-
Default combine rule of a cluster:
One combined RNA defined by the generic family of the cluster, the maximal positions, the common strand. -
IF cluster of tRNA THEN IF no tRNAscan-SE predictions THEN default combine rule ELSE IF one tRNAscan-SE prediction THEN combined RNA has the tRNAscan-SE family name, the min and max positions of the cluster for start and end positions, and the common strand ELSE (several tRNAscan-SE predictions) do nothing (do not combine the cluster) -
IF cluster of 5s_rRNA, 8s_rRNA, 16s_rRNA, 18s_rRNA, 23s_rRNA or 28s_rRNA THEN IF no RNAmmer predictions THEN default combine rule ELSE IF one RNAmmer prediction THEN combined RNA has the RNAmmer family name, the min and max positions of the cluster for start and end positions, and the common strand ELSE (several RNAmmer predictions) do nothing (do not combine the cluster) -
IF cluster of unknown THEN first, remove user added predictions second, cluster the remaining predictions considering their gene finder set (homology search, conservation anlysis, ab initio) and apply the default combine rule on each subset.
| Generic family names | Family names given by software |
|---|---|
| 16s_rRNA | 16s_rRNA, 16S_rRNA_helix_18, 16S_rRNA_helix_18, |
| 23s_rRNA | 23s_rRNA, 23S/28S_rRNA_sarcin_loop, 23S/28S_rRNA_sarcin_loop, |
| 5_8S_rRNA | 5.8S_ribosomal, 5_8S_rRNA, |
| 5s_rRNA | 5s_rRNA, Archae_5S_rRNA, Bacterial_5S_rRNA, Eukaryotic_5S_rRNA, 5S_ribosomal_RNA, Archae_5S_rRNA, Bacterial_5S_rRNA, Eukaryotic_5S_rRNA, 5S_ribosomal, 5S_rRNA, |
| Alpha_RBS | Alpha_RBS, Alpha_operon_ribosome_binding_site, |
| DnaX | DnaX, DnaX_ribosomal_frameshifting_element, |
| FMN | FMN, FMN_riboswitch_(RFN_element), |
| Leu_leader | Leu_leader, Leucine_operon_leader, |
| Lysine | Lysine, Lysine_riboswitch, |
| Mg_sensor | Mg_sensor, Magnesium_Sensor, |
| RNaseP | RNaseP, Bacterial_Rnase_P_class_A, Bacterial_Rnase_P_class_B, Bacterial_Rnase_P_class_A, Bacterial_Rnase_P_class_B, RnaseP_nuc, RnaseP_bact_a, RnaseP_bact_b, RnaseP_arch, RNaseP_nuc, RNaseP_bact_a, RNaseP_bact_b, RNaseP_arch, |
| S15 | S15, Ribosomal_S15_leader, |
| SRP | SRP_bact, SRP_RNA_Domain_IV, |
| SraA | SraA, sraA_Hfq_binding, |
| SraB | SraB, sraB_Hfq_binding, |
| SraD | SraD, sraD_Hfq_binding, |
| SraG | SraG, sraG_Hfq_binding, |
| SraH | SraH, sraH_Hfq_binding, |
| SraL | SraL, sraL_Hfq_binding, |
| Thr_leader | Thr_leader, Threonine_operon_leader, |
| Trp_leader | Trp_leader, Tryptophan_operon_leader, |
| istR | istR, istR_Hfq_binding, |
| me5 | me5, RNase_E_5'_UTR_element, |
| mini-ykkC | mini-ykkC, mini-ykkC_RNA_motif, |
| snoRNA-CD | Pyrococcus_C/D_box_small_nucleolar, Small_nucleolar_RNA_sR1, Small_nucleolar_RNA_sR2, Small_nucleolar_RNA_sR4, Small_nucleolar_RNA_sR5, Small_nucleolar_RNA_sR7, Small_nucleolar_RNA_sR8, Small_nucleolar_RNA_sR9, Small_nucleolar_RNA_sR10, Small_nucleolar_RNA_sR11, Small_nucleolar_RNA_sR12, Small_nucleolar_RNA_sR13, Small_nucleolar_RNA_sR14, Small_nucleolar_RNA_sR15, Small_nucleolar_RNA_sR16, Small_nucleolar_RNA_sR17, Small_nucleolar_RNA_sR18, Small_nucleolar_RNA_sR19, Small_nucleolar_RNA_sR20, Small_nucleolar_RNA_sR21, Small_nucleolar_RNA_sR22, Small_nucleolar_RNA_sR23, Small_nucleolar_RNA_sR24, Small_nucleolar_RNA_sR28, Small_nucleolar_RNA_sR30, Small_nucleolar_RNA_sR32, Small_nucleolar_RNA_sR33, Small_nucleolar_RNA_sR34, Small_nucleolar_RNA_sR36, Small_nucleolar_RNA_sR38, Small_nucleolar_RNA_sR39, Small_nucleolar_RNA_sR41, Small_nucleolar_RNA_sR42, Small_nucleolar_RNA_sR44, Small_nucleolar_RNA_sR45, Small_nucleolar_RNA_sR46, Small_nucleolar_RNA_sR47, Small_nucleolar_RNA_sR48, Small_nucleolar_RNA_sR49, Small_nucleolar_RNA_sR51, Small_nucleolar_RNA_sR52, Small_nucleolar_RNA_sR53, Small_nucleolar_RNA_sR55, Small_nucleolar_RNA_sR58, Small_nucleolar_RNA_sR60, snoRNA-CDbox, |
| snoRNA-HACA | Archae H/ACA sno RNA by Fabrice Leclerc, HACA_sno_Snake, |
| tRNA | tRNA-Phe, tRNA-Val, tRNA-Leu, tRNA-Ile, tRNA-Cys, tRNA-Trp, tRNA-Arg, tRNA-Ser, tRNA-Ser, tRNA-Ala, tRNA-Pro, tRNA-Thr, tRNA-Tyr, tRNA-Asp, tRNA-His, tRNA-Asn, tRNA-Met, tRNA-Gly, tRNA-Sup, tRNA-Glu, tRNA-Gln, tRNA-Lys, tRNA-Ind, tRNA-Pseudo, tRNA-SeC(p), Type_I_tRNA, Type_II_tRNA, tRNA_Bacteria, tRNA_Eukaryote, tRNA_Archaea, tRNA, |
| yybP-ykoY | yybP-ykoY, yybP-ykoY_element, |
Explore page: visualize, explore and export results
The Explore page gives an overview of all putative RNAs of the
current project, for all runs. The precise history of the project is accessible by clicking on the link history. This gives the list of all gene finders that have been runned with the exact command line, and all actions performed by the user.

You can further explore these results, and filter displayed putative RNAs, visualize them with various genome browser/viewer, merge, edite, align them and finally export them.
You can further explore these results, and filter displayed putative RNAs, visualize them with various genome browser/viewer, merge, edite, align them and finally export them.
Filtering putative RNAs
You can apply successive filters on the
list of displayed results with the filter form. To define a
filter: choose a criterion, a comparison relation, and give a
value, then click on the
update button at the end of the line. The list of
predicted RNAs in the table of results is automatically
updated. If you apply several filtering criteria, only
predictions matching all your queries will be displayed.

Special characters available:
The search is not case-sensitive.
Here are some examples of queries.
| | | OR |
| * or % | any sequence of characters (wild card) |
| ? or _ | any single-character (wild card) |
| ^ | beginning-of-line |
| $ | end-of-line |
Here are some examples of queries.
| Field | Operator | Value | |
| start | › | 1000 | putative RNAs beginning after the position 1000 |
| size | ‹ | 500 | putative RNAs whose size is lower than 500 nucleotides |
| run | != | r02 | putative RNAs not provided by run r02 |
| family | = | tRNA|rRNA | putative RNAs whose family name is trna or rrna in upper or lower case |
| family | = | ^rRNA* | putative RNAs whose family name begins with rrna in upper or lower case |
Visualizing putative RNAs with JBrowse
Clicking on the 'JBrowse view' tab, you can visualize
all selected putative RNAs with JBrowse, that is a portable
JavaScript genome browser with a linear visualization of genomes
[more information on Jbrowse].
Visualizing putative RNAs with CGview
Clicking on the 'CG view' tab, you can visualize
all selected putative RNAs with CGview.
CGView is able to generate high quality, zoomable maps of genomes
in a circular visual output with a SVG image (XML-based file format
for describing two-dimensional vector graphics both static and dynamic,
i.e. interactive or animated) [more information on CGview].
Listing putative RNAs
Results are listed in a table in the 'Table view' tab.
For each putative RNA, the following information is displayed:
For each putative RNA, the following information is displayed:
-
ID: putative RNA identifier.
Allowed characters: a-z, A-Z, 0-9, -, _, .
Maximum length: 6 characters -
Seq name: name of the query sequence (as provided
originally).
Allowed characters: a-z, A-Z, 0-9, -, _, .
Maximum length: 40 characters -
Family: family of the putative RNA family,
'unknown' when not known.
Allowed characters: a-z, A-Z, 0-9, _, ., ',', '
Maximum length: 20 -
Start: start position of the putative RNA on the query
sequence.
Allowed characters: numeric
Maximum length: -
Constraints: must be < end -
End: end position of the putative RNA on the query
sequence.
Allowed characters: numeric
Maximum length: -
Constraints: must be > begin -
Size: length of the putative RNA, in nucleotides.
Allowed characters: numeric
Maximum length: -
-
Strand: '+' corresponds to the same strand as the
query sequence, '-' to the opposite strand and '.' stands for
unknown strand.
Allowed characters: +, -, .
Maximum length: 1
-
Species: species of the putative RNA sequence
(as provided originally).
Allowed characters: a-z, A-Z, 0-9, -, _, .
Maximum length: 20
- Domain: domain (Archea/Bacteria/Eukaryote) of the putative RNA (as provided originally).
-
Replicon: putative RNA sequence replicon (as provided
by you).
Allowed characters: a-z, A-Z, 0-9, -, _, .
Maximum length: 20
-
Software: name of the gene finder used to identify the
putative RNA.
Allowed characters: a-z, A-Z, 0-9, -, :, .
Maximum length: ???
- Align.: number of alignments in which the putative RNA is involved
- Run: run identifier, of the form r01, r02, and so on.
Specify information displayed.
You may change fields of information displayed in the table using the
display functional element. Default value is 'Terse'.
If you select 'All', another column is added displaying the score
attached to a putative RNA by the gene finder. It is important
to be aware that only scores provided by a same gene finder are
comparable. Each program has its own rule to set or not the score.
By convention, tools that do not compute any score are assigned
a null value (0.0).
Specify the number of results displayed.
You may change the number of results to be displayed in a page of the
table using the drop-down list show. You may also indicate which
page to display using the page functional element.
Sort the results. By default, results are sorted by sequence,
and for each sequence by increasing start position. The list may be
sorted according to other criteria by clicking on the corresponding column
titles (except for the first column called All/None, used to
select and deselect entries).
See more details on a specific prediction. Click on the
hyperlinked putative RNA identifier (ID column) that you want to see.
It will display a new page containing all information concerning the
prediction. See Section Visualizing
and editing a prediction.
Acting on putative RNAs
The two frames located below the table of results allow you to
perform actions on the predictions of the table.
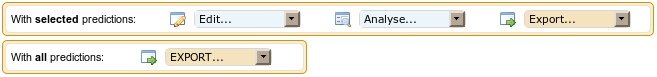
Actions from the first frame apply on a selection of putative RNAs,
whereas actions of the second frame apply on all putative RNAs present
in the table of results.
With selected predictions: Predictions can be
selected by ticking the check box in the left column of the
main table. You can also select all putative RNAs displayed on
the current page by clicking on 'All' (title of the first column).
Edit... This drop-down list allows you to modify the list of
results.
- rename family: change the family name. The new name will be assigned to all selected RNAs. It must be made up of upper and lower case letters, figures and '_' '.' '-' characters. Name length must be between 1 and 20 characters.
- split into 2 families: put selected predictions in two families. You have to specify two family names and then assign each selected putative RNAs to one of these two families. The names must be made up of upper and lower case letters, figures and '_' '.' '-' characters. Name length must be between 1 and 20 characters.
- add a new putative RNA manually: add a new putative RNA to the list of results. For that, you have to describe all features attached to the RNA: ID, family name, genomic sequence name, start position, end position, strand and optionnaly a secondary structure. The genomic sequence has to be chosen in the list of previously loaded sequences. See the section Editing a prediction for futher information concerning the other fields. Click on the save button to store the description. The new putative RNA is inserted in the pool of putative RNAs, and the Explore page is automatically updated.
- add putative RNA(s) from GFF: add new putative RNAs to the list of results. For that, you have to upload a file or paste information in GFF format. . Click on the save button to store the description. The new putative RNAs are inserted in the pool of putative RNAs, and the Explore page is automatically updated.
- merge putative RNAs: merge all selected putative RNAs into
a single prediction. The new prediction is built as following:
- ID: a new ID is created
- Start: the smallest start value
- End: the largest end value
- Strand: + if at least one prediction is +, and others are + or ., - if at least one prediction is - and others are - or ., . otherwise.
- Family: the concatenation of initial family names separated by '/' (has to be renamed if family name is longer than 20 characters)
- Software: the concatenation of software names separated by '/' and prefixed by 'Merge:'
- combine putative RNAs: combine selected putative RNAs
to reduce redundancy.
See the section Combine results for a description of the combine procedure. - delete: delete all selected putative RNAs.
Analyze... This drop-down list allows you to further analyze your
results.
- align: compare selected putative RNAs by building a multiple alignment between them. The multiple alignment is obtained with ClustalW and RNAz (see above).
- view with ApolloRNA (webstart): view selected putative RNAs
in a linear visual output with a webstart application (program is run
on the client computer) of ApolloRNA
[more information on ApolloRNA].
Note that as the software runs on the user computer, user has to first download the two files (the genomic sequence in FASTA format and the selected putative RNAs in Ensembl GFF format) provided in a ZIP file that need to be unzip. Second, user has to launch ApolloRNA and point the two local files to be loaded in ApolloRNA. Beware that for Apollo users, the local Apollo configuration will be used. In this case, the tiers for putative RNAs will not be colored in green but in light yellow (default color).
Export... This drop-down list allows
exporting selected results.
Choose the required output format, and follow the usual procedure to
save the file on your local computer.
Available export formats are
- multiFASTA
>NC_000913-RNA000032| |-|13559|13618 ACGUGCAGGAUACCGUCAGCAUCGAUAUCGAAGGUAACUUCGAUCUGCGGCAUGCCGCGCG >NC_000913-RNA000032| |-|13559|13618 ACGUGCAGGAUACCGUCAGCAUCGAUAUCGAAGGUAACUUCGAUCUGCGGCAUGCCGCGCG ... - GFF
NC_000913 blast ncRNA 228758 228817 61.3 + . ID=000001 NC_000913 blast ncRNA 228818 228872 61.3 + . ID=000002 ... - GFF for Apollo Genome Annotation
Curation Tool
Cobalamin_000162 ncRNA ncRNA 4979 5181 0.0 + . SSU_rRNA_5_000185 ncRNA ncRNA 8251 9338 0.0 + . ... - CSV (Comma-Separated Values), a very simple format supported by almost all spreadsheets (including Microsoft Excel) and database management systems
000001;sample;tRNA-Glu;9979;10054;76;+;E.coli;bacteria;Chromosome;tRNAscan-SE;59.8;1;r01 000002;sample;tRNA-Thr;16995;17070;76;+;E.coli;bacteria;Chromosome;tRNAscan-SE;91.83;0;r01 ... - RNAML with extensions. RNAML is a standard XML syntax for exchanging RNA information [more information on RNAML]. Extensions are described in the RNAspace development documentation.
With all predictions: actions available in this frame concern
all predictions of the table of results, even those non visible on the
current page of the table.
EXPORT... This drop-down list allows exporting all
predictions. It works the same way as the previously described
'Export...' drop down
list.
Visualizing and editing a prediction
On this page, you can access all information available concerning a given
prediction.
You can also modify some of it using the edit button at the bottom
of the page.
Visualizing a prediction
RNA features:The same information as displayed in the
table of
results of the Explore page.
Genome context: Flanking regions of the putative
RNA, 35 bp upstream and 35 bp downstream the prediction.
Sequence and structure: The sequence of the putative RNA is given
here.
When available, a secondary structure is also displayed in
bracket-dot format.
A base pair between bases i and j is represented by an
opening parenthesis at position i and a closing paranthesis
at position j.
Unpaired bases are represented by dots. The lack of pseudoknots in the
secondary structure ensures that this notation defines a unique folding.
ggggcuauagcucagcugggagagcgccugcuuugcacgcaggaggucugcgguucgaucccgcauagcuccacca
(((((((..((((........)))).(((((.......))))).....(((((........))))))))))))...
Several secondary structures can be attached to a single putative RNA.
For each structure, a 2D drawing is created with RNAplot
[more information on RNAplot]
or VARNA [more information on VARNA].
Alignments: The number of alignments in which the putative
RNA is involved. Alignments may come from database homology search
(see Section
Sequence homology search),
comparative analysis
(see Section comparative analysis
gene finders).
They can also have been constructed from a set of selected predictions
using the Align option of the drop-down list Analyze in
the Explore page. You can visualize the alignments by clicking on the
hyperlink.
Editing a prediction
This page lets you modify features attached to the RNA, and to
add or modify a secondary structure. Temporary results can be viewed
on the preview page. You must save all your modifications to confirm
them before returning to the Explore page (save button at the
bottom of the page).
Modifying RNA features
When the strand of the RNA is modified, the genome context and the sequence are automatically updated. The secondary structures and the alignments, if existing, are deleted.
- ID: identifier, made up of upper and lower case letters, figures and '_' '.' '-' characters. The length must be between 1 and 6 characters.
- family name: made up of upper and lower case letters, figures and '_' '.' '-' characters. The length must be between 1 and 20 characters.
- start position: must range between 1 and the last position of the query sequence.
- end position: must range betwen start+1 and the last position of the query sequence.
- strand: must have a value in '+' (same strand as the input sequence), '-' (reverse strand) and '.' (unknown).
When the strand of the RNA is modified, the genome context and the sequence are automatically updated. The secondary structures and the alignments, if existing, are deleted.
Add a secondary structure. It is also possible to provide
new secondary structures (several structures can be attached to
a single RNA prediction).
A secondary structure should be typed or pasted in the dedicated text area in bracket-dot format.
Alternatively, if the length of the sequence is neither too short nor too long, some software are provided to predict secondary structures. RNAfold [more information on RNAfold] may be used to calculate the minimum free energy secondary structure. RNAshapes [more information on RNAshapes] and UNAFold [more information on UNAFold] may predict several structures. Click on the add this structure in preview page button to temporary attach the structure at the RNA (the link will be recorded if the page is quit with the Save button). A 2D plot of the structure is then created with RNAplot [more information on RNAplot] or VARNA [more information on VARNA].
A secondary structure should be typed or pasted in the dedicated text area in bracket-dot format.
Alternatively, if the length of the sequence is neither too short nor too long, some software are provided to predict secondary structures. RNAfold [more information on RNAfold] may be used to calculate the minimum free energy secondary structure. RNAshapes [more information on RNAshapes] and UNAFold [more information on UNAFold] may predict several structures. Click on the add this structure in preview page button to temporary attach the structure at the RNA (the link will be recorded if the page is quit with the Save button). A 2D plot of the structure is then created with RNAplot [more information on RNAplot] or VARNA [more information on VARNA].
Frequently Asked Questions
How long the results are kept on rnaspace.org website ?
Results are available during 14 days.
Results are available during 14 days.
Why the execution time of a gene finder with same
parameterization could be quite different ?
The time is dependent on the charge of the server. You will be informed by mail when the execution is completed.
The time is dependent on the charge of the server. You will be informed by mail when the execution is completed.
Why do I not see the Help page when clicking on the help icon ?
The help page is opened only once, and reused. The display is changed when different items are required. When the page (may be a tab) is recalled, it is not always put at the foreground level. To do so, you have to select it explicitly.
The help page is opened only once, and reused. The display is changed when different items are required. When the page (may be a tab) is recalled, it is not always put at the foreground level. To do so, you have to select it explicitly.